Why choose exome sequencing?
The advent of whole-genome sequencing (WGS) ushered in a whole new world for biological research. Since the Human Genome Project was (partially) completed 20 years ago, the cost of WGS has plummeted, now sitting at $1000 or less (vs more than $1,000,000 at its height). However, sequencing a whole human genome is data-heavy and can take 100GB to upwards of 1TB of hard drive space for a single sequencing run, generating a vast amount of data to analyze. Enter the exome.
Whole exome sequencing (WES) sequences all the protein-coding regions (exons) of the genome but avoids the non-coding introns. Coming into vogue in the 2010s, the technique has increased exponentially in popularity in the past decade.1 There are several advantages to WES over other technologies. The exome is only about 1.5-3% of the genome, so focusing on the exome rather than the genome substantially reduces the size and cost of a sequencing run. WES runs come in at a scant few gigabytes of hard drive space, and while the cost is not 1.5-3% of WGS, WES generally costs around a quarter of a full genome.2 Exome sequencing is highly efficient, with diagnostic yields ranging from 20-40% in the clinic,3 compared with yields in the single digits for comparative genomic hybridization arrays (aCGH). Though specific figures vary from study to study, exome sequencing tends to beat microarray-based sequencing methods and targeted next-generation sequencing (NGS) gene panels by a factor of 3-4x in terms of diagnostic yield.4 When using NGS techniques to find pathogenic variants, exome sequencing shines, since 89% of variants reported as pathogenic in NCBI’s ClinVar come from protein-coding regions (at least in Mendelian disorders). That number skyrockets to 99% when neighboring regions are included.5
Starting with exome sequencing can shorten diagnostic time and increase diagnostic yield.
In a diagnostic situation, exome sequencing offers many advantages over targeted panels. A targeted panel is often selected from a phenotype-first point-of-view, where a patient’s clinical presentation guides the selection of a specific set of genes to examine. This approach can, however, be unsuccessful, resulting in either needing a whole new panel or reflexing to exome sequencing. Exome sequencing is the preferred method of diagnosis in several common scenarios: when patients have generalizable symptoms that could have arisen from a variety of underlying conditions; when a disease is genetically heterogeneous or has substantial phenotypic variability (like hydrops fatalis); or if a rare disease has a genetic cause that simply lies outside of the known genes included in targeted panels.6,7 What’s more, starting with exome sequencing can speed up diagnostic time, increasing diagnostic yield by more than 40% in one clinic’s hands. Shortening the time from presentation to diagnosis can radically reduce clinical costs and improve patient outcomes, by quickly pinpointing treatment and care options.
Covering all the bases – Achieving comprehensive coverage
Peculiarities in sequence features can be a problem for any kind of exome sequencing analysis. Both disproportionately high GC or TA content can decrease the accuracy of exome sequencing.8 Why this should be the case is not always particularly clear, though blame commonly falls on PCR or polymerase-based issues (due to the higher melting temperature for GC-rich regions for instance). Careful probe design can help avoid this problem. It can also help attenuate the challenge of capturing sequences located within highly ordered, difficult structures — particularly approaches that use overlapping probes rather than end-to-end or even gapped probes.9
Copy number variations (CNVs), mutations that duplicate or delete segments of DNA, arise in a variety of diseases. Until recently, WES was not the preferred method for CNV detection, since chromosomal microarrays performed better (with a yield of around 20% for chromosomal aberrations reported in 2010)10. WES datasets have, in the past, lacked the consistency between methods, and the high-quality reference material needed for some clinical applications. However, recent work proposed changing the order of workflow and incorporating sequencing data from outside WES databases,11 making WES a powerful technique for detecting pathogenic CNVs.
Needle in the haystack — Variant filtering and prioritization
For a wide variety of rare diseases such as metabolic, neurological, and developmental disorders, the precise causative genetic mutation has often only ever been seen once – 69.6% of cases in Orphanet (a rare disease database) have only one documentation.12 Many rare diseases can arise from a variety of contributing genes along a signaling pathway. In these situations, past efforts like those using traditional Sanger sequencing can often fail to pick up a causative variant, but there is a litany of examples of exome sequencing succeeding in their stead.13,14
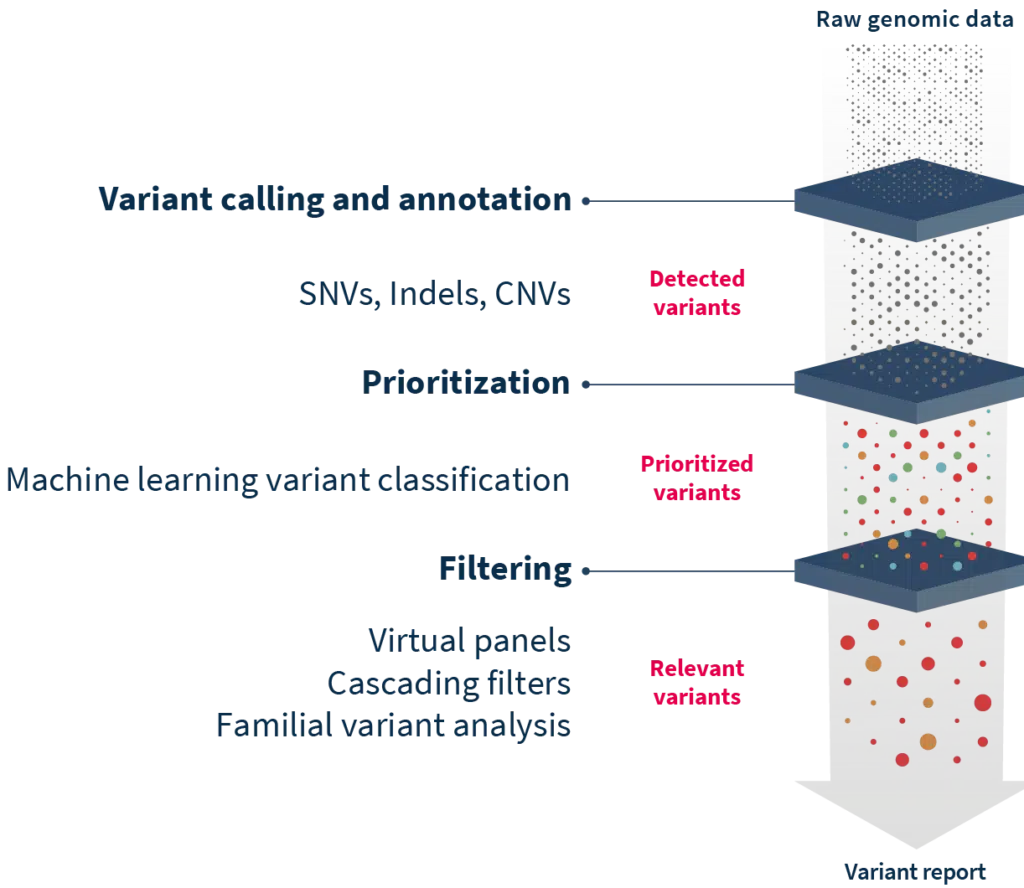
Although exome data requires heavy processing before analysis, the researcher does not necessarily require a bioinformatics skill set. Much of the processing, like trimming low-quality bases at the end of reads or detecting and deleting adapters, can be achieved automatically using specialized analytic software.
Determining which variants are pathogenic and which are not can be a difficult task. The drawback, of course, of WES versus a targeted gene panel, is that there are much more data to deal with. WES runs commonly find tens of thousands of variants – in extreme cases, hundreds of thousands.15,16 This can introduce an order of magnitude of noise into a dataset. Applying a few key rules and variant filtering strategies can reduce the number of variants by 90-95% so that researchers might then be faced with 150-500 candidate mutations, a much more manageable number.16 Further investigation requires consulting scientific databases and repositories to determine whether the detected exonic variants are pathogenic, benign, or variants of unknown significance (VUS). Unfortunately, there is no single, all-encompassing database that contains all the information needed to interpret variants. That means, unless researchers have access to software that taps into tens of different databases, it will take substantial time to manually check through one by one. In addition, despite the vast amount of information available, a significant proportion of detected variants are still VUS, highlighting the ever-growing need for more research in the field.
Exome analysis with SOPHiA DDM™ and Alamut™ Visual Plus
The SOPHiA DDM™ Platform for rare and inherited disorders accurately detects a range of variant classes, with high sequence coverage uniformity even in complex and GC-rich regions. Where multiple combinations of NGS technologies can introduce artifacts and inconsistencies, the SOPHiA DDM™ Platform filters the noise and bias to deliver advanced analytical performance independent of the input. This high-quality, noise-filtered output is used to accurately detect CNVs with exon-level resolution. The platform even covers ∼200 variants in non-coding regions and the entire mitochondrial genome to ensure comprehensive exome analysis. No matter the variant type — SNV or CNV — SOPHiA GENETICS’ analytics provide optimized variant detection in a single experiment.
Where pinpointing pathogenic mutations can be difficult, SOPHiA DDM™ complemented by Alamut™ Visual Plus can help cut through the noise and enable a deep exploration of variants. Together, the two technologies annotate variants with information from more than 55 world-renowned biological databases and repositories, including missense and splicing predictors. The SOPHiA DDM™ Platform offers additional filtering features such as Virtual Panels to limit interpretation to genes associated with specific disorders, Cascading Filters to reduce analysis to variants with specific characteristics, and Familial Variant Analysis for consideration of parental samples and inheritance mode. For on-the-ground level accuracy, users benefit from becoming members of the SOPHiA GENETICS Community, where experts flag variant pathogenicity to improve interpretation, even of VUS. Finally, for efficient and user-friendly interpretation, Alamut™ Visual Plus enhances the visualization of variants in a comprehensive full genome browser.
Innovations in sequencing and analysis aren’t slowing, and researchers need tools to keep afloat in the flood of data. Analytical technologies like SOPHiA DDM™ and Alamut™ Visual Plus are ideal parts of a researcher’s arsenal to find pathogenic variants and get clear answers from complex WES datasets.
References
- Samuels DC, et al. Trends Genet 2013;29:593-9.
- The Cost of Sequencing a Human Genome. National Human Genome Research Institute. https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost
- Clark MM, et al. npj Genomic Medicine 2018;3:16.
- Martinez-Granero F, et al. npj Genomic Medicine 2021;6:25.
- Barbitoff YA, et al. Sci Rep 2020;10:2057.
- Norton ME, et al. Am J Obstet Gynecol 2022;226:128.
- Sawyer SL, et al. Clin Genet 2016;89:275-84.
- Chilamakuri CSR, et al. BMC Genomics 2014;15:449.
- Warr A, et al. G3 (Bethesda) 2015;5:1543-50.
- Miller DT, et al. Am J Hum Genet 2010;86:749-64.
- Rajagopalan R, et al. Genome Med 2020;12:14.
- Wakap SN, et al. Eur J Hum Genet 2020;28:165-73.
- Weedon MN, et al. Am J Hum Genetics 2011;89:308-312.
- Xia X, et al. PLoS One 2016;11:e0156981.
- Carson AR, et al. BMC Bioinformatics 2014;15:125.
- Gilissen C, et al. Eur J Hum Genet 2012;20:490-7.