Menu
Unleashing the power of healthcare data with the New Generation SOPHiA DDM™ Platform.
As the burden of cancer and rare diseases continues to grow globally, the complexity of the diseases demands more sophisticated solutions. Researchers and clinicians are constantly striving to develop novel, more effective therapies, and diagnostic tools to improve patient outcomes and resolve the biggest unmet needs in global healthcare. At the core of these efforts, there is one key element: Data.
From diagnosis to therapy selection and drug development, data is now indispensable for diagnosis and personalized treatment. The rise of precision medicine highlights the critical need for cutting-edge solutions that can harness and analyze vast amounts of healthcare data, driving advanced decision-making to improve patient outcomes at scale.
In response to this pressing need, platforms like SOPHiA DDM™ have emerged as revolutionary solutions in advancing data-driven medicine. Since its initial release in 2015, SOPHiA DDM™ has pioneered how healthcare professionals use data, having analyzed over 1.8 million genomic profiles to date and accelerating the practice of precision medicine worldwide. With nearly 30,000 analyses per month, the SOPHiA DDM™ Platform has proven itself to be a vital tool in the fight against cancer, rare and inherited diseases. Yet, as healthcare evolves, so too must the tools and technologies that support it.
In recent years, the healthcare landscape has witnessed a dramatic increase in both the volume and complexity of data. Genomic, radiomic, and clinical data have become integral to understanding diseases on a deeper level. However, the ability to process, integrate, and analyze these diverse data sources remains a significant challenge for clinicians and researchers. This challenge highlights an unmet need in global healthcare: the necessity for platforms that can break silos within and among healthcare institutions, and bridge the gap between data generation and actionable insights, allowing for more accurate diagnoses and personalized treatment strategies.
In response to this emerging need, SOPHiA GENETICS has just revealed the New Generation SOPHiA DDM™ Platform, aiming to stay at the forefront of precision medicine and address today the healthcare needs of tomorrow.
The New Generation SOPHiA DDM™ Platform not only enhances the speed and efficiency of data processing but also offers a powerful, web-based architecture designed to meet the evolving demands of clinical research. By leveraging advanced technologies like cloud computing and GPUs from world-class industry partners such as NVIDIA and Microsoft, SOPHiA DDM™ is set to revolutionize how healthcare professionals manage and interpret complex datasets to make informed decisions.
Leveraging the groundbreaking capabilities of the SOPHiA DDM™ Platform, healthcare professionals benefit from significantly reduced turnaround times, enabling quicker insights from data upload to final analysis.
Moreover, the platform’s enhanced computing capabilities allow it to process larger and more complex datasets, paving the way for new applications such as Whole Genome Sequencing (WGS), Minimal Residual Disease (MRD), Liquid Biopsy, and more, providing deeper insights into the genetic underpinnings of diseases, helping clinicians tailor therapies to individual patients with greater precision.
In addition to genomics, the platform offers advanced multimodal analytics, which are essential for understanding diseases like cancer, where multiple data types (genomic, radiomic, and clinical) need to be integrated for a more comprehensive view of the patient’s condition and unique biology. This multimodal approach allows for the analysis and interpretation of diverse data across different modalities, leading to more accurate predictions and personalized treatment plans.
One of the most significant advancements in the New Generation SOPHiA DDM™ Platform is its ability to offer genomic, radiomic, and multimodal analyses within a single, integrated workspace. This unified approach empowers healthcare providers to select the tools and applications that best suit their needs, whether they are focused on identifying genetic mutations, analyzing medical images, or integrating various data sources for predictive modeling.
By integrating these diverse data types, the New Generation SOPHiA DDM™ empowers clinicians to make better-informed decisions, improving the precision of diagnosis and treatment in oncology, rare and inherited diseases.
One of the key challenges in modern healthcare is the fragmentation of data. In many systems, vital information is siloed across different platforms and institutions, limiting the ability to generate a comprehensive understanding of a patient’s condition. SOPHiA GENETICS addresses this issue by promoting a decentralized, technology-agnostic, global platform where data can be securely shared among users, breaking down barriers to knowledge and experience exchange.
As Dr. Zhenyu Xu, Chief Scientific Officer at SOPHiA GENETICS, explains, “Our decentralized, multimodal analytics platform supports customers and helps break data silos by creating a global community where knowledge is safely and securely shared amongst users. The new generation of our SOPHiA DDM™ Platform is revolutionizing the user experience by blending our powerful AI algorithms with multimodal data to produce meaningful insights to further the field of precision medicine.”
As cancer therapies and data technologies continue to evolve, platforms like SOPHiA DDM™ will play a central role in shaping the future of precision medicine. The need for innovative, data-driven solutions is more urgent than ever, as healthcare providers strive to keep pace with the complexities of modern diseases.
Abhimanyu Verma, Chief Technology Officer at SOPHiA GENETICS, reflects on the broader impact of these advancements: “We pride ourselves on adapting our technology to meet our customers’ needs. As the technology infrastructure at most healthcare organizations worldwide has evolved, we are thrilled to continue to provide best-in-class technology and set them up for success. This new generation of our platform will allow us the flexibility to respond quickly to our customer’s evolving needs and introduce new features faster, and more efficiently.”
With its innovative architecture and advanced analytics capabilities, the new SOPHiA DDM™ Platform represents a major leap forward in precision medicine. By addressing the unmet needs in global healthcare data analysis, SOPHiA GENETICS is helping to pioneer a future where data-driven insights lead to more personalized, effective, and timely care for patients around the world.
Learn more about the New Generation SOPHiA DDM™ Platform here. Interested in getting a free demo of the Platform? Book it here!
What is cascade testing?
Cascade testing is the practice of offering genetic testing to relatives of known carriers of pathogenic variants associated with autosomal dominant conditions. In oncology, cascade testing is performed in families affected by hereditary cancer syndromes1. The most common include hereditary breast and ovarian cancer syndrome (HBOC), Lynch syndrome (LS), familial adenomatous polyposis syndrome, hereditary pancreatic cancer syndrome and gastric cancer syndrome2,3. Testing for variants associated with HBOC and LS belongs to the so-called Tier 1 testing, i.e., genomic applications, the implementation of which is supported by robust evidence4.
Cascade testing starts with first-degree relatives (parents, siblings, children) of index cases (i.e., the family member in whom a pathogenic variant was identified) and then proceeds to second- (grandparents/grandchildren, aunts/uncles, nieces/nephews, half-siblings) and third-degree relatives (great-grandparents/great-grandchildren, first cousins)1.
Most hereditary cancer syndromes follow the autosomal dominant inheritance pattern. Therefore, the first-, second- and third-degree relatives have, respectively, a 50%, 25%, and 12.5% probability of inheriting the predisposition to develop cancer (see Figure 1)1. For some pathogenic variants of genes associated with hereditary cancer syndromes, such as BRCA1/2, the penetrance is high5. Establishing accurate estimates of penetrance and relative risk for genes implicated in hereditary cancer syndromes is an ongoing task3.
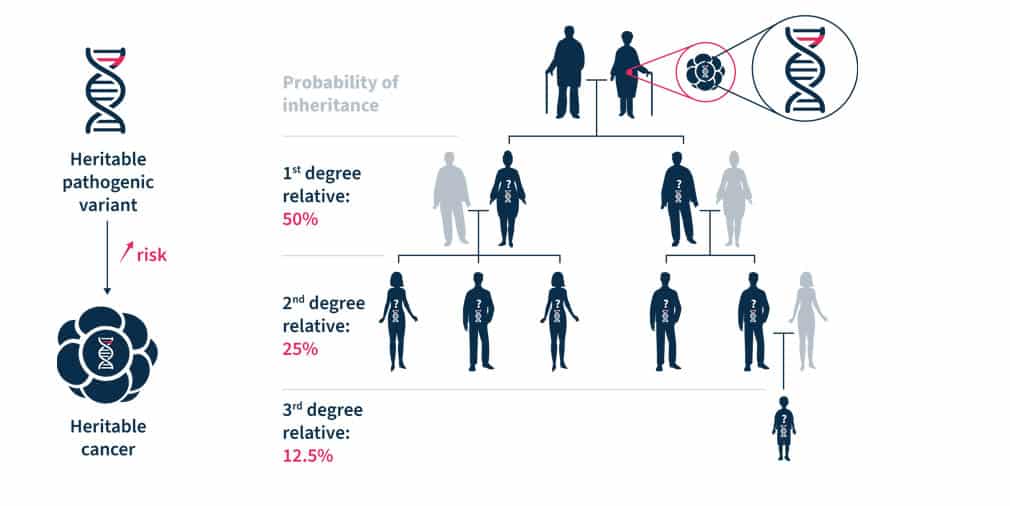
Figure 1. Heritable pathogenic variants increase the risk of developing cancer at a younger age (left). Cascade genetic testing is the practice of testing the relatives of known carriers (right)1.
Why is cascade testing important?
At the level of an individual and their family, cascade testing has two important goals. The first goal is to identify relatives that carry the familial pathogenic variant and require personalized cancer risk management1. The second goal is to exclude the non-carriers from intensive cancer surveillance and prevention interventions1. The detection of pathogenic variants in individuals at a reproductive age may lead to decisions of assisted reproduction or prenatal diagnosis. In the case of actionable monogenic conditions, cascade testing may reduce adverse health outcomes in cohorts of relatives1.
At the societal level, cascade testing has important clinical and research implications for oncology. It can further our knowledge of hereditary cancers and is a cost-effective way of identifying unaffected individuals at-risk, thus, providing important information to plan long-term resources necessary to cope with hereditary cancers. Moreover, today’s testing is needed to tailor future approaches in cascade testing1.
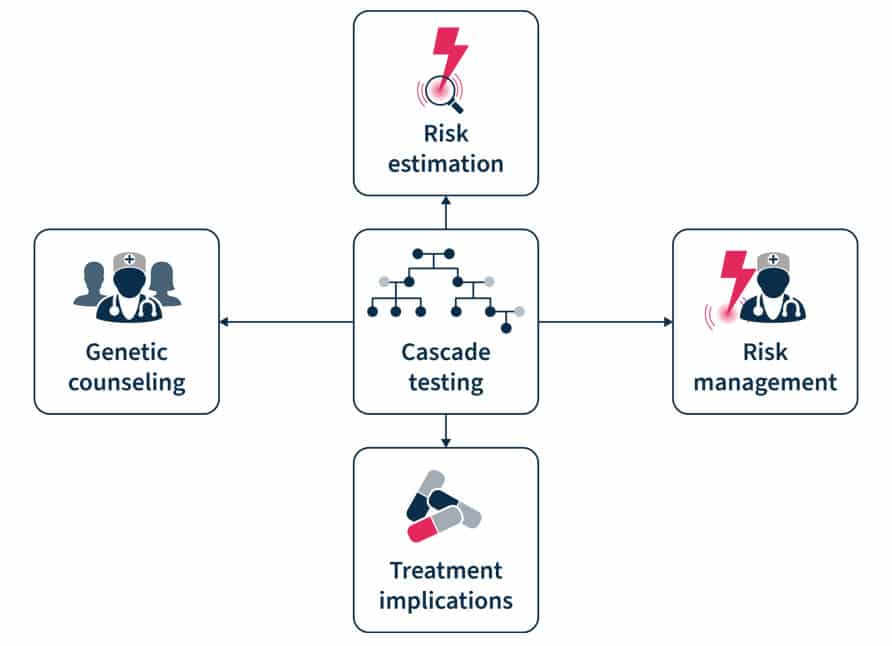
Figure 2. Cascade testing involves genetic counseling before and after the test, risk estimation and management, and has treatment implication7.
What are the barriers to cascade testing?
Despite the advantages of cascade testing, its uptake is low. The reported rates of uptake of cascade testing in HBOC and LS equals ~50% and the underutilization of testing results in missed opportunities of cancer prevention1. In a recent Swiss study, there was a 25-50% response rate to invitations to cascade testing and at least one-in-three individuals at risk did not undergo testing. An index case possesses an average of 10 relatives eligible for testing, while the average rate of genetic tests per index case is only 1.51.
There are several barriers to cascade testing6,7. These include ineffective family communication of genetic risk information, low knowledge of cascade testing among index cases and primary care providers, and geographic barriers to receiving genetic services. Cascade testing uptake is also lower among male than female relatives and in distant compared to first-degree relatives. A facilitator of adherence to cascade testing is the parents’ desire to understand their children’s risk6. “Dear family” letters, digital chatbots (a technology-based simulated conversations), and direct contact programs have been shown to be effective in motivating cascade testing8.
Several initiatives exist to promote cascade testing. One such enterprise is the Cascade Resources Network, an independently run, non-profit organization that offers access to genetic testing, genetic counseling, variant interpretation, screening guidelines, and forums and support. It was developed by Memorial Sloan Kettering Cancer Center (MSK) fellows, Ryan Kahn and Sushmita Gordhandas. The network was created to increase the rate of genetic testing among relatives of patients with inherited cancer risk variants to help identify cancer early in families and, ultimately, to prevent future cancers. Similarly, the Swiss Cancer Genetic Predisposition Cascade Screening Consortium was established in 2016 to foster research related to the hereditary cancer predisposition. In particular, the Consortium promotes the CASCADE cohort, a family-based open-ended cohort targeting HBOC and LS variant-harboring families to elicit factors that enhance adherence to testing (NCT03124212).
Analyze genetic predisposition to cancer with the SOPHiA DDMTM Platform
Multi-gene testing is an efficient, affordable, and guideline-recommended9 approach to cascade testing as it allows for comprehensive assessment of biologically relevant hereditary cancer genes. The SOPHiA DDM™ Platform supports various next generation sequencing (NGS)-based Hereditary Cancer Applications to help clinician researchers characterize the complex mutational landscape associated with hereditary cancer disorders.
Powered by advanced analytics, users can detect challenging variants in a streamlined sample-to-report workflow, including:
Variant pathogenicity levels are assigned using machine learning complemented by guideline-driven ranking, helping to prioritize relevant variants and reduce interpretation time. Furthermore, deeper variant exploration is supported by Alamut™ Visual Plus, a full-genome browser that integrates numerous curated genomic and literature databases, guidelines, missense and splicing predictors.
To learn more about SOPHiA DDM™ for Hereditary Cancers, explore here or request a demo here.
References
1. Sarki M, et al. Cancers (Basel) 2022;14:1636.
2. Brown GR, et al. JAAPA 2020;33(12):10-16.
3. Mighton C, Lerner-Ellis JP. Genes Chromosomes Cancer 2022;61(6):356-381.
4. Dotson WD, et al. Clin Pharmacol Ther 2014;95(4):394-402.
5. Chen S, Parmigiani G. J Clin Oncol 2007;25(11):1329-1333.
6. Roberts MC, et al. Health Aff (Millwood) 2018;37(5):801-808.
7. O'Neill SC, et al. Hered Cancer Clin Pract 2021;19(1):40.
8. Campbell-Salome G, et al. (2022) Transl Behav Med 2022;12(7):800–809.
9. Daly MB, et al. J Natl Compr Canc Netw. 2021 Jan 6;19(1):77-102.
The HGVS nomenclature guidelines are used worldwide for genetic variant interpretation but can seem complicated and difficult to understand and apply. That is why we have created this beginner’s guide to mutation nomenclature using the HGVS recommendations, with clear visual examples that break down the process into bitesize pieces.
1. What is HGVS nomenclature?
2. How to read mutation nomenclature: Breaking down the variant description
2.1 Reference sequence e.g., NM
2.2 Description of variant e.g., c.4375C>T
2.3 Predicted consequence e.g., p.(Arg1459*)
3. The 3 prime rule for mutation
4. Final thoughts and helpful tool
The Human Genome Variation Society (HGVS) nomenclature standard was developed to prevent the misinterpretation of variants in DNA, RNA, and protein sequences. The HGVS nomenclature standard is used worldwide, especially in clinical diagnostics, and is authorized by the Human Genome Organisation (HUGO).1,2
HGVS General Terminology Recommendations1
| X Do not use | ✔️ Recommended terminology |
| Mutation or polymorphism | Variant, change, allelic variant Can be used for cancer tissue: Mutation load and tumor mutation burden |
| Pathogenic | Affects function, disease-associated, phenotype-associated |
HGVS follow recognized standards for the nomenclature of DNA and RNA nucleotides, the genetic code, amino acid descriptions, and cytogenetic band position in chromosomes.3
The HGVS recommendations for mutation nomenclature state that the format of a complete variant description should first include the reference sequence, followed by the variant description, and then the predicted consequence in parentheses. For example, NM-004006.2:c.4375C>T p.(Arg1459*) (Figure 1).

The HGVS nomenclature recommendations for sequence variants state that a complete variant description should begin with the reference sequence.1 The reference sequence accession number begins with a two-letter abbreviation (explained in Table 1), followed by a multi-digit number, and finally a version number.
Table 1. Meaning of the two-letter abbreviation at the beginning of a reference sequence accession number.
| Abbreviation | Reference sequence based on a: |
| NC | Chromosome |
| NG | Gene or genomic region |
| LRG | Locus Reference Genomic sequence: Gene or genomic region, used in a diagnostic setting |
| NM | Protein-coding RNA (mRNA) |
| NR | Non-protein-coding RNA |
| NP | Protein (amino acid) sequence |
The variant description begins by depicting the type of reference sequence used (c = coding DNA sequence, g = genomic reference sequence). When a protein-coding reference sequence is used (c), the nucleotide numbering begins with a 1, which represents the first position in the protein-coding region (the A of the translation-initiating ATG), and ends at the last position of the stop codon. Thus, if you divide the position number by 3, you can identify the affected amino acid in the protein sequence e.g., using the same example as above, 4375/3 = 1459, indicating that the predicted consequence affects amino acid 1459, which is an arginine. Different variants are indicated using different notations (explained in Table 2).
Table 2. HGVS notation and examples for the most common types of mutations2
| Notation | Example | Explanation |
| > | c.4375C>T | Substitution of the C nucleotide at position c.4375 with a T |
| del | c.4375_4379del or c.4375_4379delCGATT | Nucleotides from position c.4375 to c.4379 deleted |
| dup | c.4375_4385dup or c.4375_4385dupCGATTATTCCA | Nucleotides from position c.4375 to c.4385 duplicated |
| ins | c.4375_4376insACCT | ACCT inserted between positions c.4375 and c.4376 |
| delins | c.4375_4376delinsACTT or c.4375_4376delCGinsAGTT | Nucleotides from position c.4375 to c.4376 (CG) are deleted and replaced by ACTT |
When only DNA has been analyzed, the RNA- and protein-level consequences of the variant can only be predicted, and should thus be reported in parentheses e.g., p.(Arg1459*) is the predicted effect at protein level (p) for the example described above.
For all variant descriptions using HGVS nomenclature, the nucleotide at the most 3’ position of the variation in the reference sequence is arbitrarily assigned to have changed (see how to apply this rule in Figure 2).4 The exception is for deletions/duplications around exon junctions for which shifting the variant 3’ would place it in the next exon.5

Although the HGVS recommendations can be difficult to understand and might take a bit of getting used to, if you break them down and refer to the examples in this guide, you are on the road to success!
If you want to accelerate your variant annotation and interpretation, Alamut™ Visual Plus is a comprehensive, full genome browser for efficient and user-friendly variant interpretation. The software accelerates the complex and time-consuming assessment of variants thanks to its user-friendly interface and integrated features for variant annotation and analysis.
Find out how Alamut™ Visual Plus applies the HGVS nomenclature recommendations to ensure that variant annotation follows the universally applied standards for variant analysis, interpretation, and reporting in our dedicated Technical Note.
Alamut™️ Visual Plus is for Research Use Only. Not for use in diagnostic procedures.
References
Dr. Mohamed Z Alimohamed kindly summarized his upcoming peer-reviewed publication in Gene:
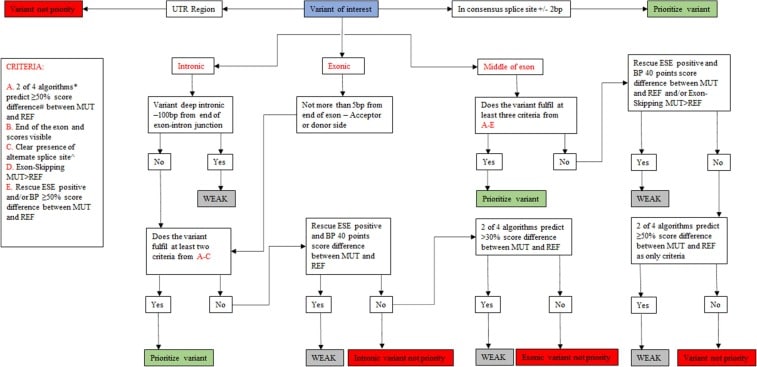
“Current splice prediction algorithms have limited sensitivity and specificity, therefore many potential splice variants are classified as variants of uncertain significance (VUSs).
However, functional assessment of VUSs to test splicing is labor-intensive and time-consuming. We have developed a decision tree, SEPT-GD, by setting thresholds for the splice prediction programs implemented in Alamut™️ to prioritize potential splice variants associated with cardiomyopathies for functional studies, and functionally verified the outcome of the decision tree.
SEPT-GD outperforms the tools commonly used for RNA splicing prediction and improves prioritization of variants in cardiomyopathy genes for functional splicing analysis.”
Click here to read the full publication.
Alimohamed MZ, Boven LG, van Dijk KK, Vos YJ, Hoedemaekers YM, van der Zwaag PA, Sijmons RH, Jongbloed JDH, Sikkema-Raddatz B, Westers H. SEPT-GD: A decision tree to prioritise potential RNA splice variants in cardiomyopathy genes for functional splicing assays in diagnostics. Gene. 2023 Jan 30;851:146984.
Alamut™️ is for Research Use Only. Not for use in diagnostic procedures.
Discover what makes SOPHiA DDM™ effective at calling mtDNA variants from exome sequencing data, and see for yourself how in a single workflow, the SOPHiA DDM™️ Platform complemented by Alamut™️ Visual Plus can be used to identify and interpret variants in mtDNA alongside SNVs, Indels, and CNVs in nuclear DNA.
[divi_shortcode id="10875"]
Our Technical Note outlines the guidelines and standards behind the nomenclature convention deployed in Alamut™ Visual Plus. We also explain how Alamut™ Visual Plus applies them to ensure that variant annotation follows the universally applied standards for variant analysis.
[divi_shortcode id="10869"]
At the American Society of Human Genetics (ASHG) Annual Meeting this year, our esteemed speakers shared the ins and outs of how the SOPHiA DDM™ Platform, in combination with Alamut™ Visual Plus, adapted to their laboratories’ needs to provide sample-to-report workflows that streamlined the identification and interpretation of nuclear and mitochondrial variants associated with rare and inherited diseases, including hereditary cancers.
All SOPHiA GENETICS™️ products discussed in this article are for Research Use Only – not for use in diagnostic procedures. SOPHiA GENETICS™ does not facilitate and does not accept any liability for any validation of SOPHiA GENETICS™ products for clinical use by a third party.
Exome sequencing with combined mitochondrial genome sequencing for the detection of nuclear and mitochondrial DNA variants
Jessica Van Ziffle, PhD, FACMG Associate Clinical Professor, Pathology at University of California, San Francisco, California, United States
In the Pathology laboratory at the University of California, the proportion of positive/probably positive variants detected in pediatric and prenatal cases analyzed using the SOPHiA DDM™ Custom Whole Exome Solution (optimized for the Illumina NovaSeq 6000) was consistent with the literature (see charts). Approximately 70% of the positive pediatric cases were associated with autosomal dominant inheritance. Most reported variants were missense single nucleotide variants (SNVs), with the positive cases fairly equally split between frameshift, nonsense, and missense variants.
Proportion of positive/probably positive findings for pediatric and prenatal exome cases
Figure sourced from Jessica Van Ziffle’s presentation
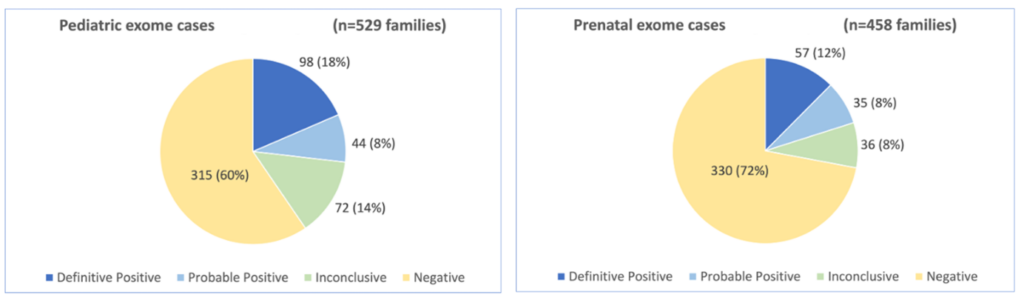
With the goal of increasing the proportion of positive findings, Jessica and the team at UCSF explored what additional variants could be identified by exome sequencing to potentially solve the ∼10% of inconclusive cases and ∼65% of negative cases. First, the team investigated the impact of calling copy number variants (CNVs), specifically contiguous gene changes, whole gene changes, and exon-level changes. UCSF worked closely with SOPHiA GENETICS™ to optimize their exome sequencing to ensure high and even coverage for accurate CNV detection, and indeed doubled their sequencing depth to 80M reads to ensure the sensitive detection of CNVs 1-2 exons in size.
Next, UCSF wanted to be able to simultaneously call variants in mitochondrial DNA, which is especially relevant for metabolic diseases. Different cell types have different numbers of mitochondria, and each mitochondrion has its own genome that can have different variants in it (heteroplasmy). The UCSF team, therefore, wanted to assess the lower limit of detection for mitochondrial heteroplasmy through a mixing study, which concluded that exome testing could detect variants down to 5% variant allele fraction with high sensitivity.
Streamlining clinical implementation of hereditary cancer analysis and reporting with a custom application
Hong Wang, PhD, FCCMG, FACMG, DABMGG Laboratory Geneticist at North York General Hospital, Toronto, Ontario, Canada
Andrea Vaags, PhD, FCCMG Discipline Co-Lead and Laboratory Geneticist at Trillium Health Partners – Credit Valley Hospital, Mississauga, Ontario, Canada
Drs Wang and Vaags provided a step-by-step overview of how they developed a brand new hereditary cancer panel to meet the Ontario Health - Cancer Care Ontario criteria for hereditary cancer testing.
Laboratory and clinical working groups were established to evaluate evidence and identify key genes and non-coding variants to include in a cutting-edge custom hereditary cancer panel. Furthermore, genetic testing eligibility criteria were co-developed with the Hereditary Cancer Clinical Eligibility Working Group. The laboratory working group used an evidence-based framework to design a standardized 76-gene panel, organized into 13 larger disease site-linked panels, and 25 single/small gene panels. After designing the panel, the Ontario group worked with SOPHiA GENETICS™ to expeditiously develop and implement the custom SOPHiA DDMTM Hereditary Cancer application in academic community hospitals (see timeline).
Timeline of SOPHiA DDM™ Hereditary Cancer panel implementation in Ontario hospitals
Figure sourced from the Ontario hospital responsible for validating this product
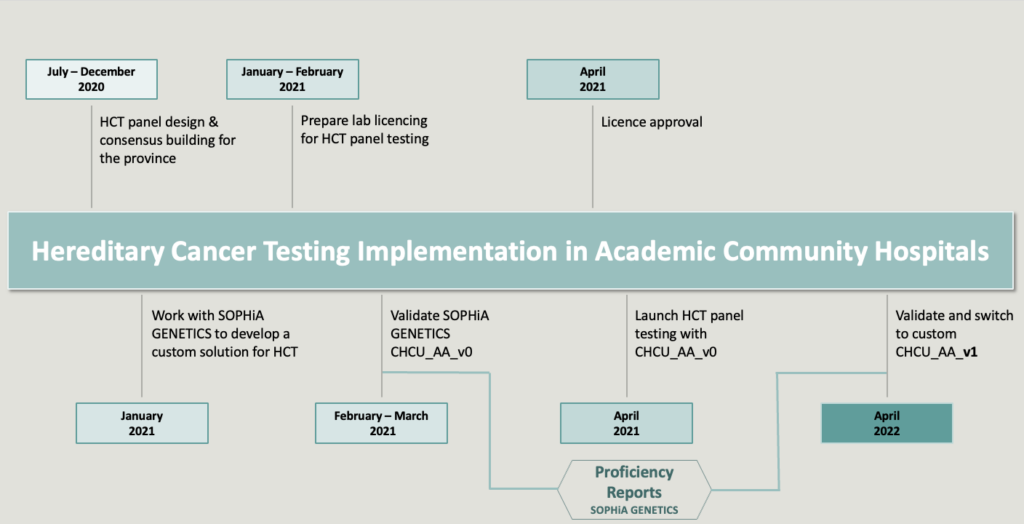
Thanks to the streamlined end-to-end SOPHiA GENETICS™ workflow, hereditary cancer testing approximately doubled, according to Dr Vaags.
The workflow for each batch of 70 samples (plus one control) in the Ontario group laboratories, consists of DNA preparation, automated 3-day library preparation using the SOPHiA GENETICS program on the Hamilton STARlet, and sequencing on a NextSeq® 550 using mid-output. Sequencing data are automatically uploaded to the SOPHiA DDM™ cloud for processing ahead of analysis. For additional time savings, genes requiring special consideration due to the presence of pseudogenes are flagged with a warning in the SOPHiA DDM™ Platform and the SOPHiA GENETICS™ support team is on hand to answer queries on unusual findings. The cloud-based software for data management enables the laboratories to streamline data access, storage, and archiving back-up. Dr Wang shared that the time saved through this workflow has been instrumental in maintaining turnaround times, especially with significant understaffing during challenging periods.
By applying Virtual Panels and custom filters, the teams can analyze from as little as a single variant to as many as 76 genes using a single workflow. The high analytical sensitivity and specificity enable the laboratories to pick up unusual findings, such as Alu insertions, Boland inversions, and low-level mosaicism of copy number changes. And finally, the one-step secondary and tertiary analysis for concurrent detection of SNVs and CNVs allows the teams to significantly speed up their analysis, and the pseudogene pipeline enables the laboratories to minimize reflex testing. In summary, the custom SOPHiA DDM™ Hereditary Cancer application provides the Ontario laboratories with a one-size-fits-all solution.
Screening for genetic variants in hereditary cancer syndromes using the end-to-end SOPHiA DDM™ workflow
Mark Williams, FHGSA – Chief Scientist at Genomic Diagnostics, Heidelberg, Victoria, Australia
Speaker Mark Williams began his talk by highlighting that a key goal of the Genomic Diagnostics lab is to facilitate equal access to hereditary cancer testing. To do this, the lab set multiple criteria that were highly important to them when developing a new hereditary cancer application. Employing the complete SOPHiA GENETICS™ workflow for Hereditary Cancer allowed the team at Genomic Diagnostics to successfully meet these testing criteria.
In collaboration with Genomic Diagnostics, the custom SOPHiA DDM™ Hereditary Cancer application was designed to include genes that align with current practice guidelines. It was important to the lab that the pipeline could detect SNVs, Indels, and copy number variations (CNVs) in a single workflow. Mark confirmed that the resultant application effectively detects CNVs and is scalable, with turnaround times that meet their needs, even with testing volumes increasing year-on-year. In addition, the solution provides high-quality, consistent results, a full record of curation, visualization of BAM files, and is easily accessible and usable by all laboratory staff.
Like numerous other SOPHiA GENETICS™️ customers, Mark concluded that the SOPHiA DDM™ Platform offers a robust, automated, and secure bioinformatics pipeline that meets Australia’s privacy regulations. In addition, the software is extremely user-friendly, from its visual interface to the detailed QC metrics, annotation information, and links to databases. All laboratory personnel can effectively use the end-to-end solution, even without prior bioinformatics expertise.
The integrated workflow and affordable price allowed Genomic Diagnostics to expand access to the custom SOPHiA DDM™ Hereditary Cancer application (see chart), meeting Genomic Diagnostics’ goal of facilitating equal access to hereditary cancer testing.
Increasing access to hereditary cancer genomic testing over time
Figure sourced from Mark Williams’ presentation
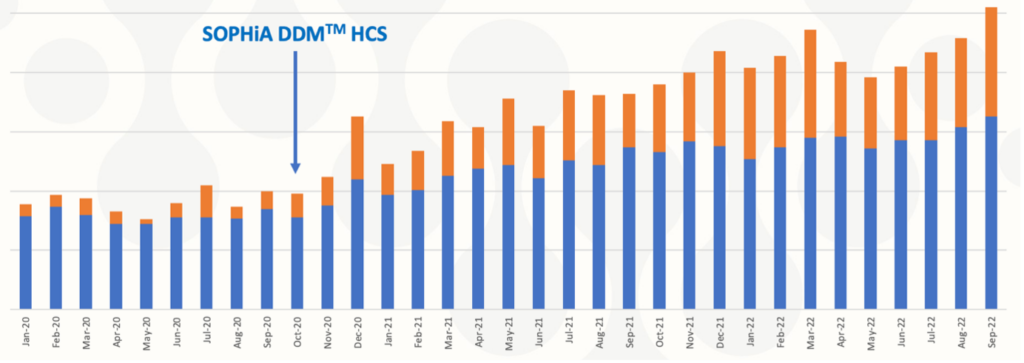
We thank all our speakers for sharing their research stories at our ASHG symposium this year. We’re delighted to hear how their integrated SOPHiA DDM™ workflows are reducing workloads, expanding access, and continuing to discover new variants associated with rare diseases and hereditary cancers.
SOPHiA GENETICS™ celebrated a significant milestone, reaching 1 million genomic profiles analyzed by our data analytics platform SOPHiA DDM™. While this certainly marked a meaningful achievement for us as a company, it has even greater significance for the accuracy of our machine-learning pipelines.
Our decentralized model has generated data in over 70 countries, adding diversity to our database and enabling the platform to identify variants not present in other reference databases. Nearly 19% of valuable variants identified by SOPHIA DDM™ are yet to be identified in other databases. We asked some of our internal experts how this exposure to such a vast genomic data set impacts our algorithms.
First, we chatted with Emily Paul, PhD, a Product Director on our Platform team, leading enhancements to SOPHiA DDM™. Emily expressed that the 1 million genomic profiles milestone is significant in three ways:
Sequencing 1 million profiles would not have been possible without our growing community of institutions embracing next-generation sequencing (NGS). As the access to NGS testing increases, sharing knowledge worldwide also becomes possible. With 1 million profiles sequenced, the platform has seen exponentially more variants, increasing the possibility of identifying those with previously unknown significance through community flagging. This large amount of data provides our machine learning algorithms with an additional source of information, allowing them to identify variants that would not traditionally be flagged. This milestone increases the platform’s exposure to a vast number of variants. It strengthens the power of SOPHiA DDM™,leading to better detection and enhanced classification of rare variants, which can result in better patient outcomes.”
We were also able to catch up with Zhenyu Xu, PhD, Chief Scientific Officer at SOPHiA GENETICS™. We asked him what the 1 million profiles mean for the platform and how valuable he sees this data.
These 1 million genomic profiles capture the bias inherent to different NGS technologies and library preparation methodologies, as well as the diversity of ethnicities and diseases. Together, they give our algorithms access to a wealth of information, allowing for a more comprehensive understanding of the relationship between genetic diversity and pathology. They also empower us to build more robust cohorts for future research. As a result, we have fine-tuned our pipelines for more precise analyses and can now pinpoint individual profiles for our customers faster and more efficiently.
We are proud of this accomplishment and excited about how we can use this information to create more accurate analytical pipelines to support your research. You can learn more about SOPHiA DDM™ here.
Familial Hypercholesterolemia (FH), is the most common monogenic autosomal dominant disorder where affected individuals present with significantly elevated low-density lipoprotein (LDL) cholesterol in the blood, tendinous xanthomas, corneal arcus, and coronary artery disease (CAD)1. This affects about 1 in every 200-250 people globally, but most people are unaware they have it2, 3.
FH is known to be caused by inherited mutations in genes used to regulate and remove cholesterol in the blood. Among the individuals with a clinical diagnosis of FH, pathogenic variants can be identified in one of the four genes - LDLR, APOB, LDLRAP1, and PCSK9 in about 60-80% of adult and 60-95% of pediatric FH patients1, 2, 4. Other known genes implicated in FH are – ABCG8, ABCG5, APOE, and LIPA5, 6.
There are two types of FH – Heterozygous FH (HeFH) and Homozygous FH (HoFH). HeFH is caused by a single inherited variant from one parent. In rare cases, an individual can have HoFH, which is caused by having two causal variants inherited from each parent. Individuals with HoFH typically have a more severe form of the disease. LDL receptors usually remove LDL-C from the blood to the liver by carrying the lipoproteins that fix LDL-C for transport. Genetic mutations in the LDLR gene can cause a decrease in the number of LDL receptors or interfere with its normal functions. This causes the high LDL cholesterol levels seen in FH patients.
FH remains underdiagnosed and undertreated globally. Additionally, patients with HoFH are diagnosed much later and are at higher premature Atherosclerotic Cardiovascular Disease (ASCVD) risk.
FH can be diagnosed using targeted molecular testing or next-generation sequencing (NGS) gene panel strategies; however, the latter is still not widely used6, 7. In 20-30% of individuals that meet clinical criteria for FH, standard clinical genetic testing may be negative due to either technical limitations (e.g., clinical sensitivity of current technology) or causal genes that are not yet discovered. FH is diagnosed in children with LDL‐C persistently over 160 mg/dL (4.1 mmol/L) and adults with LDL‐C greater than 190 mg/dL (4.9 mmol/L), especially if there is a family history of early‐onset CAD, and in all patients with early CAD.
The results of population-based studies of genetic screening for FH have demonstrated that there is no fixed LDL-C threshold for making the diagnosis of HoFH8. Although an untreated LDL-C of over 400 mg/dL (10 mmol/L) should lead to a consideration of the diagnosis of HoFH, LDL-C levels of less than 400 mg/dL have also been documented in patients with genetically confirmed HoFH. These high cholesterol levels can lead to a variety of symptoms. Cholesterol build-up can deposit in different parts of the body which can lead to deposits around the elbows and knees, Achilles tendon pain, and or a white or grey ring around the iris of the eye. FH is an inherited disease present from birth, but symptoms may not manifest until adulthood, which makes more early testing necessary.
Universal screening for FH is currently not feasible or cost-effective. The most effective means to identify new cases of FH is by cascade screening family members of a known index case8. Cascade genetic testing is beneficial in developing early intervention strategies for affected family members. Screening of relatives can be done by measuring LDL-C, genetic analysis, or both, and FH has been designated as a tier 1 genomics application for family screening by the US Centers for Disease Control and Prevention Office of Public Health Genomics. Universal pediatric screening coupled with reverse cascade screening has been proposed as another strategy to identify new FH cases as the discrimination power between FH and non-FH cases based on LDL-C levels is better during childhood8.
Despite the lack of screening, FH disorders are easily actionable. Patients with FH can have an excellent prognosis once the disorder is diagnosed, and a treatment plan is put in place. Dietary and lifestyle modifications are the starting points for LDL‐C lowering in patients with FH, but multidrug treatment is often required to achieve adequate LDL‐C levels. The risk of developing CAD is increased up to 13-fold in untreated FH subjects, and 22-fold when an FH mutation is present.
Statins are potent competitive inhibitors of 3-hydroxy-3-methylglutaryl coenzyme-A reductase and have proven useful in the treatment of FH. It is recommended to treat FH patients with high doses of high‐intensity statins, which are capable of lowering LDL‐C by 50% to 60%8. If high‐dose, high‐intensity statins are not tolerated, the maximally tolerated statin dose is prescribed. PCSK9 inhibitors such as inclisiran (small interfering RNA) and evolocumab, (human monoclonal antibody), can also be used to lower lipid levels. Lipoprotein apheresis is indicated in HoFH or severe heterozygous FH patients with inadequate response to cholesterol-lowering therapies. Lastly, significant global disparities exist in treatment regimens, control of LDL cholesterol levels, and cardiovascular event-free survival, which demands a critical re-evaluation of global health policy to reduce inequalities and improve outcomes for all patients with HoFH.
Studies show that as few as 10% of individuals living with FH could be aware of their diagnosis7. Consequently, FH is most often diagnosed in adulthood after a cardiac event. The widespread access to genetic testing could be part of the solution, but concerted efforts are still necessary to raise awareness of FH and identify barriers to comprehensive screening, early diagnostics, and treatment.
Whole exome sequencing (WES) is one of the latest advancements in next-generation sequencing (NGS) technology.This method sequences about 233,785 exons, or protein coding regions of the genome. This region is about 20,000 genes which makes up only about 1 percent of the human genome. Mutations in these regions can alter the respective proteins leading to various phenotypic implications. Analyzing the whole exome can unravel causative variants for diseases ranging from Mendelian to complex phenotypes.
When to use whole exome sequencing versus targeted or whole-genome sequencing
Targeted or panel sequencing only sequences genes that are known to be associated with a disease. It is often used, if the given disease has a clinical testing panel available and if the focus is solving the case with a short turnaround time.
However, if disease mechanisms are poorly understood, it can take several gene panels to identify the putative variant. Thus, WES allows for all protein-coding genes to be analyzed at once, which increases the probability of identifying the causative variant in a single sequencing assay. Since WES is not restricted to evaluating genes that have previously been associated with a specific disease, it gives a more comprehensive overview of the exome. Additionally, WES can help identify novel disease-gene associations in a research setting.
Another type of NGS is whole-genome sequencing (WGS), which evaluates the entire genome. This method can also be valuable for diseases with complex phenotypes or for cases where large structural variations are the primary cause of the disease. WGS can identify large structural variations and splicing variants in deep intronic regions. However, the large volumes of sequencing data pose some unique challenges for data analysis and storage. Although genomic sequencing continues to become cheaper and more accessible, it can be cost restrictive for some institutions.
Hence, WES can present a Goldilocks option between targeted gene panels and WGS. It generates a more comprehensive genomic view than targeted panels, but creates a data volume that is more manageable to analyze than that generated by WGS. In some specific cases, it also gives the researcher an opportunity to analyze copy number variations (CNV). Lastly, WES has a higher throughput and faster turnaround time compared to WGS.
Specific patient types who could benefit from whole exome sequencing
WES is an effective tool that can be used in a multitude of situations, but there are a few specific situations in which it presents significant advantages. One such situation is when an individual has overlapping phenotypes across multiple rare diseases. Using WES and taking a genotype-first approach can identify causal disease-gene associations and help diagnose overlapping conditions. This more comprehensive view can also shorten turnaround time for diagnosis compared with running a series of targeted panels.
The shorter turnaround time and higher diagnostic yield with WES also presents significant advantages for neonatal patients. Phenotypes can be difficult to assess in neonatal patients and some symptoms might not yet manifest. Comprehensive genetic testing such as WES can help to reach an early diagnosis, which not only allows for the implementation of an optimized care plan but can also provide information on any preventative measures and/or other potential complications that could arise as a result of the disorder.
Rare disease diagnosis is often an exhaustive journey for patients and family members seeking answers for their accurate diagnosis and disease management through several specialists, while undergoing multitude of tests and procedures, in pursuit of receiving the most effective treatment. WES can be a good follow-up to those initial tests, providing information that could uncover relevant variants in genes that were not previously implicated in a given disease (diagnosis).
Thus, WES cannot only be used for identifying causative variants related to the patient’s disorder, but the comprehensive approach can provide information about additional diseases or any genetic predispositions to other inherited disorders. This information is sometimes known as secondary or incidental findings and allows for patients to better manage their condition and understand the associated health risks. In some cases, this information may not be reported back to the patient, however, having the sequenced exomes allows for easier reanalysis of the data and future germline assessment of the same patient.
Considerations for implementing whole exome sequencing
A major logistical consideration for implementing WES beyond (the cost of) validating a new test is choosing which bioinformatics platform will be used to analyze the data. As touched on previously, WES produces much more data than sequencing using a targeted panel. Most whole-exome solutions sequence around 20,000 genes and a targeted panel that may sequence less than 100 genes. This larger volume of data is responsible for the advantages of WES, by creating a more comprehensive view of the genome which can increase diagnostic yield. Institutions that are interested in bringing on WES need to consider the large volume of data being generated and how this bioinformatic workflow can be challenging for finding relevant variants.
A bioinformatics platform for WES analysis should accurately and reliably identify multiple variant types in a single workflow and have a fast turnaround time and multiple filtering options to help streamline variant interpretation. The SOPHiA DDM™ Platform delivers advanced analytical performance, can complete WES analysis overnight, and has dedicated filtering features and a rich knowledgebase to help identify variants of interest associated with rare diseases for research purposes.
SOPHiA GENETICS products are for Research Use Only and not for use in diagnostic procedures unless specified otherwise.
SOPHiA DDM™ Dx Hereditary Cancer Solution, SOPHiA DDM™ Dx RNAtarget Oncology Solution and SOPHiA DDM™ Dx Homologous Recombination Deficiency Solution are available as CE-IVD products for In Vitro Diagnostic Use in the European Economic Area (EEA), the United Kingdom and Switzerland. SOPHiA DDM™ Dx Myeloid Solution and SOPHiA DDM™ Dx Solid Tumor Solution are available as CE-IVD products for In Vitro Diagnostic Use in the EEA, the United Kingdom, Switzerland, and Israel. Information about products that may or may not be available in different countries and if applicable, may or may not have received approval or market clearance by a governmental regulatory body for different indications for use. Please contact us at support@sophiagenetics.com to obtain the appropriate product information for your country of residence.
All third-party trademarks listed by SOPHiA GENETICS remain the property of their respective owners. Unless specifically identified as such, SOPHiA GENETICS’ use of third-party trademarks does not indicate any relationship, sponsorship, or endorsement between SOPHiA GENETICS and the owners of these trademarks. Any references by SOPHiA GENETICS to third-party trademarks is to identify the corresponding third-party goods and/or services and shall be considered nominative fair use under the trademark law.